

V-SYNTHES: A New Digital Tool for Drug Development
BY MICHAEL BORCHARD
Problems in Novel Drug Development
Drug development is an extremely costly and time consuming process. It is estimated that on average, bringing a new treatment from concept to market costs $2.6 billion dollars and takes over 12 years,1 with only 12% of products making it through clinical trials.2 This makes it clear that there is great motivation to streamline the process in any way possible.
Most drugs work by binding to protein targets, called receptors, in or around a cell. Investigating new drugs has typically been done by screening a physical library of existing compounds for a potential match to a known receptor, testing them one by one in real cell cultures, and further modifying the promising prospects to maximize a factor called binding affinity. A high binding affinity is important because it makes a drug more likely to act upon a receptor and ultimately produce the desired effects. The difficulty of drug design is maximizing this parameter, which effectively results in a more efficient drug.
Recent advances in computational chemistry have produced digital libraries of billions of compounds, which can be run through programs to test binding to different receptors. However, these databases are lacking in diversity and the programs have a limited computing capacity, which “slows the pace of drug discovery, usually yielding initial hits with modest affinities [and] poor selectivity.”3
A New Solution
A new technology, developed by researchers at the USC Dornsife College of Letters, Arts and Sciences and the UNC School of Medicine, appears to address these issues. V-SYNTHES – virtual synthon hierarchical enumeration screening – is a computational method that requires a fraction of the computational resources while still screening libraries of compounds thousands of times faster than before and with a success rate over twice as high.
How V-SYNTHES Works
Figure 1. Red: Scaffold; Green, Blue, Purple: Synthons. These chemical building blocks combine to fit the shape of a target receptor.
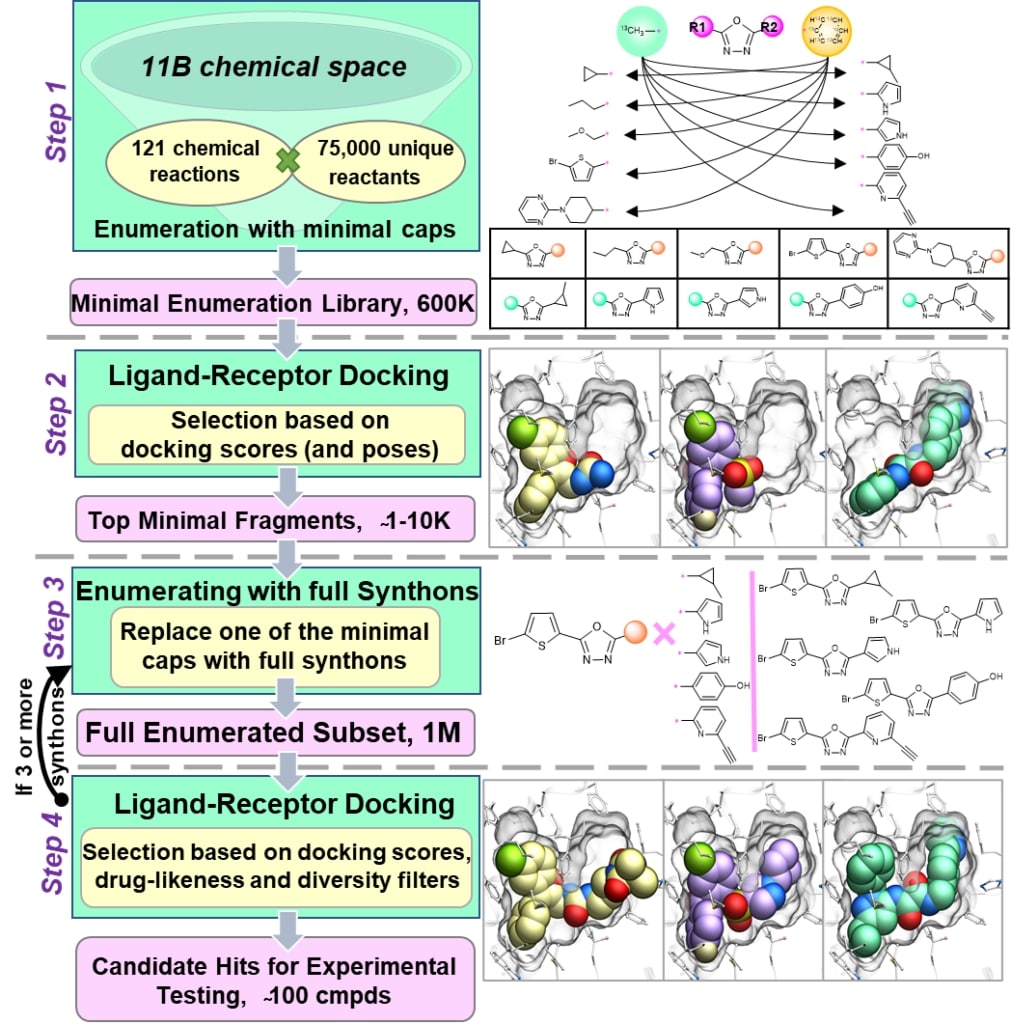
Figure 2. Summary of the V-SYNTHES process. Credit: Vsevolod Katritch, Ph.D. https://katritch.usc.edu/
How does this new method work? In their study published in Nature,3 the researchers referenced what is known as the REAL Space chemical library. Developed by the pharmaceutical company Enamine, this database “comprises 21 billion make-on-demand molecules”, which are all formed from a given set of synthons, or chemical building blocks.4 These chemical building blocks are designed to be attached to a chemical base, or scaffold. V-SYNTHES begins by creating all possible scaffold-synthon pairs from the library (~600,000)3, and assessing their binding affinity to a target receptor. This is done by simulating each molecule’s fit to a well characterized 3-D structure of the given receptor. The top scoring compounds (a few thousand) are further modified by attaching another synthon, and this set goes through another round of binding affinity testing. This is repeated once more, resulting in a final set of compounds that consist of a scaffold with three synthons attached (Figure 1). The selection process shows why V-SYNTHES is described as hierarchical enumeration – each additional synthon adds structure to the previous compound that may make it an even better fit to the receptor. The top ranked compounds at the end undergo post processing, “filtering for physico-chemical properties, drug likeness, novelty and chemical diversity,” to get a final set of 50-100 candidates for actual synthesis and experimentation (Figure 2).3 With this method, only a small subset of the library needs to be analyzed at each step, greatly reducing the computational cost.
Implementation of V-SYNTHES in Drug Development
The initial study of V-SYNTHES used the CB2 cannabinoid receptor as a template and effectively screened 11 billion compounds from the REAL Space.3 The hierarchical synthon selection process allowed the program to analyze <0.1% of the possible compounds, reducing computational resources by a factor of 5,000. Of the final compounds tested, 33% showed a sufficiently high binding affinity, compared with 15% using previous methods, and the compounds showed high selectivity, “with only negligible off-target activities.”3 Three of the top ranking compounds were then used as references to search for similar compounds in the REAL Space, which yielded 60 analogues with very high potency. The advantage of V-SYNTHES was replicable with other proteins with similar results. V-SYNTHES is already changing the way that drug development is done. As researchers continue to improve computational methods and expand the possible virtual chemical space, what used to take years of toil in the laboratory can happen virtually in a matter of weeks. This new method is a leap forward for the initial phases of drug development – it is faster, more accurate, applicable to likely any protein target, and adaptable to different databases. Bryan L. Roth, MD, PhD, a coauthor of the study from UNC School of Medicine explains the importance of the new method; “V-SYNTHES represents a major advance in the field of drug discovery. It is easily scalable and adaptable, and it should open new vistas in the discovery of potentially therapeutic chemicals for a large number of disorders at a rate never before possible.”6 Chemical space is massive, and efficient tools like V-SYNTHES will be needed to address the ever growing need for new therapeutics.
References
- Mohs, R. C.; Greig, N. H. Drug Discovery and Development: Role of Basic Biological Research. Alzheimer’s & Dementia: Translational Research & Clinical Interventions 2017, 3 (4), 651–657., https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5725284.
- Austin, D.; Hayford, T. Research and development in the pharmaceutical industry. https://www.cbo.gov/publication/57126 (accessed Feb 23, 2022).
- Deane, C.; Mokaya, M. A Virtual Drug-Screening Approach to Conquer Huge Chemical Libraries. Nature 2021, 601 (7893), 322–323., https://www.nature.com/articles/d41586-021-03682-1.
- Real space. https://enamine.net/compound-collections/real-compounds/real-space-navigator (accessed Feb 23, 2022).
- Joy, D. S. Now scientists can efficiently screen billions of chemical compounds to find effective new drug therapies > news > USC dornsife. https://dornsife.usc.edu/news/stories/3599/now-scientists-can-efficiently-screen-billions-of-chemical-compo/ (accessed Feb 23, 2022).
- Derewicz, M. Scientists unveil drug discovery tool to screen more than 11 billion compounds. https://news.unchealthcare.org/2021/12/scientists-unveil-drug-discovery-tool-to-screen-more-than-11-billion-compounds (accessed Feb 23, 2022).